This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The Office of Pharmaceutical Quality (OPQ) in the US Food and Drug Administration (FDA)’s Center Pharmaceutical Quality for Drug Evaluation and Research (CDER) has released its 2022 annual report analysing drug manufacturers and their products. FDA’s sampling and testing programme found 892 of 1,552 product samples (57.5
The FDA approves new cancer treatments in half the time of the EMA – but does faster mean better? And how can regulators balance timely access with robust safety? They found that the FDA approved 85 (95%) of the drugs before the EMA, with the latter clocking up a median delay of 241 (150-370) days.
The US Food and Drug Administration (FDA) has approved Rykindo ® (risperidone) for extended-release injectable suspension. According to 2022 data gathered by the World Health Organization (WHO), there are around 24 million schizophrenia patients. Clinical trials of the FDA approved Rykindo ®.
The US Food and Drug Administration ( FDA) has approved Wezlana (ustekinumab-auub) as a biosimilar to Johnson & Johnson’s Stelara (ustekinumab). billion in 2022, according to J&J’s financial results. The post FDA approves first Stelara biosimilar, Wezlana appeared first on European Pharmaceutical Review.
UK-based pharmaceutical giant GSK has announced that the US Food and Drug Administration (FDA) has extended the review period of its new drug application (NDA) for the rare bone cancer drug momelotinib by three months. In July 2022, GSK acquired the therapy as part of its $1.9bn acquisition of Sierra Oncology.
The US Food and Drug Administration (FDA) has granted Fast Track designation for Pfizer and BioNTech’s messenger ribonucleic acid (mRNA)-based combination vaccine candidate against Covid-19 and influenza. The post US FDA grants Fast Track status for Pfizer-BioNTech’s combination vaccine appeared first on Pharmaceutical Technology.
The US Food and Drug Administration (FDA) has approved Gilead’s Sunlenca (lenacapavir), a capsid inhibitor for adults living with human immunodeficiency virus type 1 (HIV-1), who cannot be successfully treated with other available treatments. Sunlenca is given in combination with other antiretrovirals.
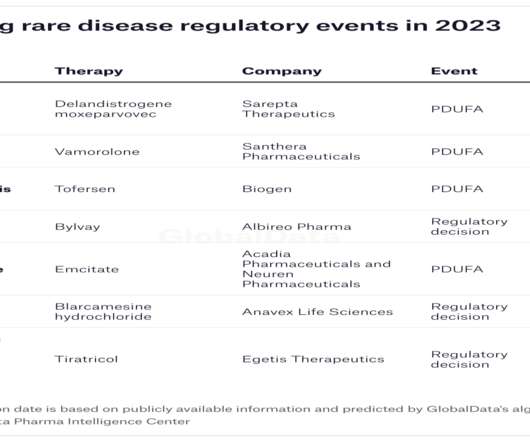
In 2022, the FDA approved only 37 new medicines, an underwhelming number compared to 98 in 2018. However, while only around 34% of the approvals in 2018 were for orphan drugs, 54% new approvals in 2022 were for drugs to treat rare diseases. The company plans to submit an MAA to the EMA in H1 this year.
The industry has been limited in its ability to implement creative solutions to engage and motivate patients because the FDA has strict guidelines for what pharma companies can communicate with patients. Measuring and monitoring adherence in clinical trials would add valuable information about the effectiveness and safety of drugs.
Food and Drug Administration (FDA) has granted an Investigational Device Exemption (IDE) to advance the company’s PATCH Clinical Study, a multi-center, single-arm, pivotal study evaluating the safety and effectiveness of the Vivasure PerQseal ® Closure Device System. The post Vivasure Medical Announces FDA IDE Approval to Initiate U.S.
This new collaboration is set to further improve safety and efficacy of radiopharmaceuticals. The US Food and Drug Administration (FDA) approved Novartis’ Pluvicto (lutetiumlutetium Lu 177 vipivotide tetraxetan) in 2022. “Radioligand therapies hold transformative potential for certain forms of cancer.”
But in early 2022, the country’s health minister announced his approval of research into psychedelic therapies and increased funding for mental health research. 3 This notice came on the heels of a January 2022 notice that clarified Health Canada’s desired approach to psilocybin research. Breakthrough therapy designation.
1 Consequently, the US Food and Drug Administration (FDA) and other agencies are keen to see “accelerated development” programmes in areas where there is a significant unmet clinical need. He is currently a CMC consultant with an interest in impurities and safety‑based limits. FDA; 2018 [cited 2023Jan]. Internet] 2022.
“The current version will enable reproducible analysis of patient data which will provide immediate value for our current and future biologics and drug delivery partners as they study volumetric infusion characteristics and longitudinal patient comparisons as part of their safety and pivotal trials. About ClearPoint Neuro. Are you hiring?
On February 7, at a town hall organised to discuss clinical trial designs for gene therapies, FDA experts pushed pharma players to look for ways to establish clinical effectiveness despite the challenges in recruiting patients with rare diseases.
This is largely due to the FDA’s rigorous approach to the safety of microbiome therapeutics, which has manifested in clinical holds, resulting in delays that have dimmed the enthusiasm in the space in recent times. While the French MaaT Pharma has submitted further information to the FDA, its trial remains on hold.
The US Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER) has accepted the Biologics License Application (BLA) for nirsevimab for the prevention of respiratory syncytial virus (RSV) lower respiratory tract infection (LRTI) in all infants. The safety profile of nirsevimab was similar to placebo.
MOMENTUM was a randomised, double-blind Phase III clinical trial of momelotinib versus danazol in symptomatic and anaemic myelofibrosis patients treated with a US Food and Drug Administration (FDA)-approved JAK inhibitor.
Following the US Food and Drug Administration (FDA)’s approval of Ferring Pharmaceuticals’ gene therapy Adstiladrin ® (nadofaragene firadenovec-vncg) in December 2022, new long-term follow up data has been revealed. The KM-estimated median duration of HG recurrence-free survival was six months, with a 30.1
has announced its B-cell maturation antigen (BCMA)-CD3-targeted bispecific antibody (BsAb) elranatamab for relapsed or refractory multiple myeloma (RRMM) has received Breakthrough Therapy Designation from the US Food and Drug Administration (FDA). The investigation noted elranatamab delivered a manageable safety profile. Pfizer Inc.
Krystal’s Phase 3 randomized, double-blind, intra-patient placebo-controlled study evaluated the efficacy and safety of B-VEC as a redosable gene therapy in DEB patients aged 6 months and older. Subjects returned to the clinical site 30 days following the last dosing visit for safety evaluation by the trial investigator.
In this article, we will explore the regulatory environment of pharmaceutical marketing and how companies are navigating it to balance patient safety and industry innovation. The United States Food and Drug Administration (FDA) is the primary regulatory agency that oversees the approval, promotion, and advertising of drugs.
Based on the results, Pfizer said it intends to submit a Biologics License Application (BLA) to the US Food and Drug Administration (FDA) for RSVpreF “in fall 2022”. Results also indicated the investigational vaccine was well-tolerated, with no safety concerns. “We g RSVpreF or placebo in a 1:1 ratio. percent was observed.
These concentration measurements are used as part of regulatory decisions regarding the safety and efficacy of medicinal products. The US Food and Drug Administration (FDA) implementation date was 7 November 2022. The draft ICH M10 guideline was published for comments in 2019.
Clinical trials for exa-cel The ongoing Phase I/II/III open-label trials, CLIMB-111 and CLIMB-121, are designed to assess the safety and efficacy of a single dose of exa-cel, the CRISPR-based medicine, in patients ages 12 to 35 years with TDT or with SCD, respectively. Costs and profits worldwide are shared with CRISPR Therapeutics.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
Sodium oxybate — which in the late 1980s was marketed to bodybuilders and then became known as GHB and criminally used as a date rape drug — has been sold under the brand name Xyrem after gaining FDA approval in 2002. In 2020, the FDA-approved indication was expanded to include those patients who suffer from cataplexy.
Although we have yet to understand the human microbiome and its role in disease fully, the scientific evidence on the efficacy of complex live biotherapeutic products (LBPs) in modulating the microbiome is striking, as evidenced by the approval of the first two commercial donor-derived products, Rebyota and VOWST, in 2022 and 2023, respectively.
The US Food and Drug Administration (FDA) has approved Dupixent ® (dupilumab) as a treatment for children patients aged one to 11 years, weighing at least 15kg with eosinophilic esophagitis (EoE). This new authorisation by the FDA’s expands its initial approval for EoE in May 2022 for patients aged 12 years and older, weighing at least 40kg.
Data announced at the European Lung Cancer Congress (ELCC) earlier this month revealed the long-term efficacy and safety of RYBREVANT ® (amivantamab) in patients with post-platinum EGFR Ex20ins-mutated advanced non-small cell lung cancer (NSCLC). 2022; 10(1). 2022; 24(2): 89 – 97. References 1. Garrido P et al.
The European Commission has followed the lead of the US FDA and approved AstraZeneca’s Tezspire as an add-on maintenance therapy for patients with severe asthma, becoming the first and only biologic that can be used in all patients, and not restricted to those with specific forms of the disease.
The report, Clinical Trials – The Importance of Diversity in Clinical Trials , examined data from clinical trials initiated between 1 January 2013 and 16 June 2022. Earlier this year the US Food and Drug Administration (FDA) published draft guidance to support companies in enrolling more ethnically diverse trial populations.
During the ‘Next-Generation Cytokine Therapy’ session at the Society for Immunotherapy of Cancer’s (SITC) 2022 annual meeting, Dr A Naing (University of Texas MD Anderson Cancer Center) presented the results from two cohorts of patients from the phase 2a clinical trial NIT-110, with relapsed/refractory (r/r) gastrointestinal indications.
The US Food and Drug Administration (FDA) has granted an Orphan Drug Designation to Editas Medicine’s gene therapy EDIT-301 in sickle cell disease, based on an April 27 announcement. The US agency previously granted the Orphan Drug Designation to EDIT-301 for its study in beta thalassemia, in May 2022. g/dL and 45.4%
The biotech said earlier it planned to seek FDA approval to start clinical trials towards the end of 2022. Initial phase 1/2a clinical findings for in CLL and multiple myeloma patients showed evidence of anti-tumour activity and an encouraging safety profile, with further readouts due before the end of the year.
The FDA has studied the importance of caregivers to drug development and regulatory decision-making. Caregiver observations of the patient experience over the progression of the disease may inform the safety and efficacy of a particular therapeutic. Accessed March 21, 2022. Accessed March 12, 2022. What Can Pharma Do?
It used to take years for a drug to progress from laboratory research to FDA approval. The FDA’s recently-launched Real-Time Oncology Review program is a good example of this modernization. The post The Latest Biotech Trends of 2022 appeared first on Impetus Digital. Faster Pharmaceutical Testing and Approvals.
The US Food and Drug Administration (FDA) has approved the first oral monotherapy treatment for adults with paroxysmal nocturnal haemoglobinuria (PNH). Over one-third of those patients require blood transfusions at least once per year, according to research published in a 2017 paper in Blood and a 2022 paper in Annals of Hematology.
On November 17, 2022, the FDA approved Provention Bio’s Tzield (teplizumab) , making it the first and only treatment to delay the onset of type 1 diabetes (T1D). The approval of Tzield, a first-in-class therapy, adds an important new treatment option for certain at-risk patients,” says an FDA spokesperson.
Overcoming challenges in patient safety, manufacturing and supply of radiopharmaceuticals As clinical trials progress and the first results are published, companies’ best candidates will emerge in the next five years. Hospital staff must also be protected from radiation while handling the agent.
Editas Medicine will release new efficacy and safety results of its gene therapy EDIT-301 in severe sickle disease as part of an oral presentation at the European Hematology Association’s (EHA) Hybrid Congress. Last month, the US Food and Drug Administration (FDA) granted the treatment an orphan drug designation for sickle cell disease.
A previous October 2020 report concluded that longer-term safety and efficacy were needed to update health-benefit price benchmarks. In August 2020, the agency had rejected an approval application, suggesting the company complete its Phase III Gener8-1 study and include two-year follow-up safety and efficacy data. million ($1.5
The US Food and Drug Administration (FDA) has gra nted approval for AbbVie and Janssen Biotech’s Imbruvica (ibrutinib) to treat chronic graft versus host disease (cGVHD) in paediatric patients aged one year and above. The post US FDA approves AbbVie-Janssen’s Imbruvica for cGVHD in children appeared first on Pharmaceutical Technology.
Food and Drug Administration (FDA) has approved MRI conditional labeling for the Evoke ® System, the first and only precision, dose-control spinal cord stimulation (SCS) therapy powered by SmartLoop technology. Specific scan conditions and safety information are provided in the Evoke ® SCS System MRI Guidelines manual. Are you hiring?
We organize all of the trending information in your field so you don't have to. Join 8,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content