This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The US Food and DrugAdministration (FDA) has approved GlaxoSmithKline ’s (GSK) Jesduvroq (daprodustat) to treat anaemia caused by chronic kidney disease (CKD) in adults who have been on dialysis for at least four months. Jesduvroq is an oral hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI). dL) for the patients.
In the US, botanical dietary supplements can be sold without US Food and DrugAdministration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few?
The Phase III RATIONALE 301 study evaluated the efficacy and safety of tislelizumab versus sorafenib as a first-line systemic treatment in participants with unresectable hepatocellular carcinoma. . HCC accounted for more approximately 85 percent of the 900,000 new liver cancer cases in 2020, according to the Globocan 2020 database. .
Overall, the authors summarised that the enhanced sensitivity improves safety assessments for volatiles in medical devices. The paper discussed the detection of volatile extractables released during the clinical use of medical devices.
Adakveo (crizanlizumab) received conditional authorisation by the EC in October 2020. However, the STAND results did not suggest new safety concerns with crizanlizumab. Crizanlizumab is approved for use by the United States Food and DrugAdministration (FDA).
The US Food and DrugAdministration (FDA) has released a new draft guidance to further support the use of decentralised clinical trials (DCTs) for drugs, biologics and devices. This draft guidance builds on recommendations published by the agency in 2020.
Pharmaceutical marketing is a critical aspect of the industry, as it helps in spreading awareness about the benefits and potential side effects of different drugs. In this article, we will explore the regulatory environment of pharmaceutical marketing and how companies are navigating it to balance patient safety and industry innovation.
To ensure the safety of medicines post-regulatory approval, a risk management plan (RMP) is established. An RMP is submitted as part of the dossier of all new drug applications and is evaluated by regulatory authorities before authorisation is given. What are risk minimisation methods? How is the efficacy of RMMs assessed?
Based on scientific knowledge and process understanding, manufacturers of drugs and drug substances have a broad awareness of potential impurities – including nitrosamine impurities – that could form during manufacturing or degradation. 1 Nitrosamines are not new or unknown impurities.
In this Q&A, Karen Pinachyan, Head of Medical Affairs Europe at CSL Behring summarises key considerations for gene therapy drug development and the ideal approach for alleviating economic strain when advancing these modern treatments. The US Food and DrugAdministration (FDA) requires a patient follow up period of at least 10-15 years.
Various uses soon became apparent including those for supply chain management, anti-counterfeiting, and to improve patient safety. In recent times, suppliers have also chosen other systems that rely on blockchain and sensors to track drug supply and reduce the influx of counterfeit drugs.
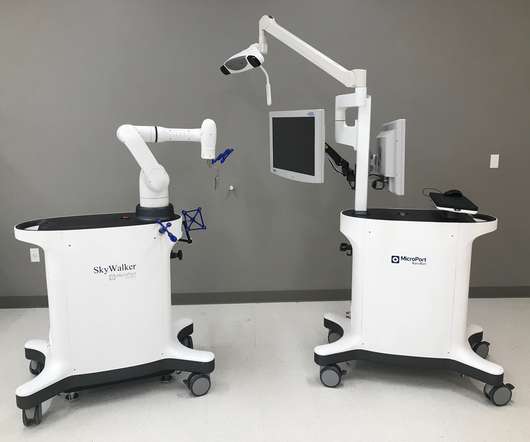
MicroPort Navibot has received 510(K) clearance from the Food and DrugAdministration (FDA) in the United States for the SkyWalker System, the company’s first robot-assisted platform for orthopedic applications. NaviBot is subsidiary of MicroPort MedBot. About MicroPort MedBot Shanghai MicroPort MedBot (Group) Co.,
In July 2020, the European Commission (EC) granted conditional marketing authorisation to bulevirtide for urgent access to HDV treatment. It has also received breakthrough therapy and orphan drug designations from the US Food and DrugAdministration.
Of the 175 new drugs approved by the US Food and DrugAdministration (FDA) between 2016 and 2019, most were biologics. When the balloon reaches a sufficient pressure, this pushes the dissolvable needle into the intestinal wall, delivering the drug payload.”
The trials analysed the safety and efficacy of daprodustat to treat anaemia of CKD in more than 8,000 patients who received the treatment for up to 4.26 Based on findings from three international Phase III clinical trials in patients from the ASCEND trial programme, the agency issued a positive opinion for the therapy.
The US Food and DrugAdministration (FDA) subsequently developed a liquid chromatography – high resolution mass spectroscopy (LC-HRMS) method for the determination of NDMA in ranitidine. 1,2 The second occurrence of NDMA arising from degradation was in metformin drug product.
Data announced at the European Lung Cancer Congress (ELCC) earlier this month revealed the long-term efficacy and safety of RYBREVANT ® (amivantamab) in patients with post-platinum EGFR Ex20ins-mutated advanced non-small cell lung cancer (NSCLC). Why are bispecific monoclonal antibody drugs such as amivantamab promising for this indication?
Notably, Lupkynis is the first oral therapy approved by the US Food and DrugAdministration (FDA) and EC for the treatment of active LN. Key opinion leaders (KOLs) interviewed by GlobalData believe this may have significant impact on patients as the less-invasive route of administration will be preferred by many.
IL-2 therapy is associated with safety concerns, namely the adverse event capillary leak syndrome, which often leads to hospitalisations and thus limits its clinical use. Protein engineering comes with several challenges, including maintaining stability and immunogenicity of the novel protein and creating an inherently active drug.
The China National Medical Products Administration’s (NMPA) Center for Drug Evaluation (CDE) has granted priority review for InnoCare Pharma ’s orelabrutinib to treat relapsed or refractory marginal zone lymphoma (R/R MZL). The therapy obtained conditional approval from the NMPA for two indications in December 2020.
Drug developers often face a Catch-22 regarding clinical trials and pregnancy. How feasible is it to establish a drug’ssafety for use in a population that may be unwilling or unable to participate in clinical trials? Panelists were asked to discuss the potential use of the drug during pregnancy.
The US Food and DrugAdministration (FDA) has accepted a priority review of the biologics license application (BLA) for Regeneron Pharmaceuticals ’ pozelimab to treat children and adults with ultra-rare CHAPLE disease. It was also granted Orphan Drug Designation during the same time.
The US Food and DrugAdministration (FDA) has approved Valneva SE’s IXCHIQ ® , the first chikungunya vaccine to be authorised in the world. In October 2023, Valneva submitted a marketing application for the chikungunya vaccine to the European Medicines Agency (EMA), which included initial safety data from the trial.
The validation of computerized systems should be focused on ‘intended use’ and scaled commensurate with the risks to patient safety, product quality and data integrity” The focus until now has too often been on compliance and not on quality. ISPE GAMP, RDI Good Practice Guide: Data Integrity by Design, ISPE, 2020. References.
We received encouraging feedback from the US Food and DrugAdministration (FDA) on the trial design and potential for accelerated approval. These efficacy results, combined with the safety profile that allows patients to complete their treatment regimen, are extremely encouraging.
In 2019, Daiichi Sankyo entered a global development and commercialisation agreement with AstraZeneca for Daiichi Sankyo’s lead antibody-drug conjugate (ADC), Enhertu (trastuzumab deruxtecan), in a deal worth $6.9bn. overall response rate (ORR), with a median duration of response (DOR) of 8.7
Information can be found in certain US pharmacopeia guide chapters, such as USP <1116>, 1 as well as guidance from the US Food and DrugAdministration (FDA) and World Health Organization (WHO) but none of these are regulatory requirements. From this observation, all options are possible.
In the current US Congressional session, Congress has focused heavily on legislation directed at reducing prescription drug prices. The new law will also require drug manufacturers to pay rebates to Medicare if they increase drug prices faster than consumer inflation. 5) monitoring access to biosimilars. Senate Bill 562 (S.
Recent policy and regulatory moves have begun to sketch out a framework for rare disease drug approvals in the country, but obstacles to approvals and patient access remain. The list, designed to serve as a reference point for prioritized drug review and evaluation policies, included 121 rare diseases. [8]. 10] [11] [12].
Lower DCT usage for Phase I studies may be accounted for due to the heavier focus on safety and efficacy, and increased monitoring of trial participants in-person. In the US, the FDA (Food and DrugAdministration) granted accelerated approvals of medical devices with AI in 2022 [8] , and growing use is expected throughout 2023.
Storage in other conditions can cause damage to the antigen or adjuvant and reduce the vaccine’s safety or efficacy. From 2020 to 2021, cases of malaria increased from 245 million to 247 million in 84 malaria-endemic countries. The majority of this rise came from the African region.
The US Food and DrugAdministration (FDA) has pushed quizartinib’s Prescription Drug User Fee Act (PDUFA) date in newly diagnosed FLT3-ITD positive acute myeloid leukemia (AML) for the review of updates of Risk Evaluation and Mitigation Strategies (REMS) included in the application. AML makes up 23.1%
Hervé Affagard (HA): Developing microbiome therapeutics as well as every other new therapeutic modality comes with several challenges that need to be addressed carefully to ensure patient safety and maximise the potential benefits of these treatments. Conversely, in the US, it has always been clear and regulated as a drug.
Adjuvanted vaccine Arexvy has been approved by the US Food and DrugAdministration (FDA). Clinical evidence that led to the FDA’s landmark approval The safety and effectiveness of Arexvy is based on the FDA’s analysis of data from an ongoing, randomised, placebo-controlled clinical study in individuals 60 years of age and older.
1 Some five years after the initial NDMA (N-nitrosodimethylamine) contamination issue 2 initially affecting valsartan drug substance, then other active pharmaceutical ingredients (APIs), eg, sartans, ranitidine, metformin, etc; the toxic short alkyl chain N-nitrosamine issue appears, if not resolved, then well on the way to resolution.
Food and Administration (FDA) approval, a change long sought by animal welfare organizations. Drugs can be now cleared for human trials using non-animal technologies developed over the last 10-15 years. Since then, animal-based testing has been the gold standard for establishing a drug’ssafety and efficacy.
They must be familiar with the different national authorities; the Food and DrugAdministration (FDA), the European Medicines Agency (EMA), and other regulations that govern the pharmaceutical marketing and sales. Department of Justice, 2020). Department of Justice, 2020).
2 The US Food and DrugAdministration (FDA) has recently announced that it will control the quality of tobacco products, particularly e-cigarettes, more closely, to prevent avoidable contamination and help address “inconsistencies between product labelling and actual concentrations” in these products, potentially misleading customers.
The success of mRNA vaccines against SARS-CoV-2 has quickly catapulted mRNA therapeutics as a disruptive, expanding drug category” The term ‘mRNA’ has become commonplace globally. The success of mRNA vaccines against SARS-CoV-2 has quickly catapulted mRNA therapeutics as a disruptive, expanding drug category.
This also occupies a large resource, given the US Food and DrugAdministration (FDA) requirement for double plate checking using a second ‘independent’ person. The US federal drug GMP regulations that first became official in 1977 and their European counterparts will need to be re-interpreted for this new generation of products.
Limitations of monoclonal antibody therapies Regulatory approvals from the US Food and DrugAdministration (FDA) for aducanumab and lecanemab – and likely very soon for donanemab also – opened a route for different therapeutic modalities and other relevant disease targets, such as tau.
The US Food and DrugAdministration (FDA) has sent a warning letter to KVK-Techs drug manufacturing facility following an inspection in April 2019. Although the particles were filtered out, the company failed to thoroughly examine their origin, nature, and potential impact on drug quality.
Merck’s old drugs are under more scrutiny, and anti-vaxxers are causing problems by cherry-picking unreliable information. regulators knew about reports of suicidal behavior in men taking the anti-baldness treatment Propecia when they decided not to warn consumers of those potential risks in a 2011 update of the popular drug’s label.
3 High attrition rates in early clinical trials have bedevilled the industry in recent years, and the average likelihood of approval (LOA) for a new Phase I drug is now just 6.7 17 AI also has useful applications in GMP manufacturing of drug substance and drug products. How Successful Are AI-Discovered Drugs In Clinical Trials?
We organize all of the trending information in your field so you don't have to. Join 8,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
































Let's personalize your content