This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Approval of Akebia Therapeutics’ Vafseo is based on additional post-marketing safety data from Japan, where the drug has been used since 2020. The FDA rejected the drug two years ago due to concerns about cardiovascular safety.
The FDA approves new cancer treatments in half the time of the EMA – but does faster mean better? And how can regulators balance timely access with robust safety? They found that the FDA approved 85 (95%) of the drugs before the EMA, with the latter clocking up a median delay of 241 (150-370) days. An abundance of caution?
Accelerated Approval allows for early access to drugs and biologics based on initial evidence of safety and effectiveness, while confirmatory studies required to verify clinical benefits are ongoing. The program was codified into law under the Food and Drug Safety and Innovation Act (FDASIA) in 2012. Do patients care?
Moderna, maker of one of only two vaccines granted emergency authorizations to prevent COVID-19 in the US, has been shamed with a 2020 “Shkreli Award” by the Lown Institute, a healthcare think tank. Can the FDA really control the quality of vaccine orders this big? However, in vaccines, cut corners could compromise patient safety.
During the first quarter of 2020, the number of telehealth visits increased by 50%, compared with the same period in 2019, with a 154% increase in visits noted in surveillance week 13 in 2020, compared with the same period in 2019. The FDA can be myopic when researching how consumers view and react to DTC.
The US Food and Drug Administration (FDA) has approved GlaxoSmithKline ’s (GSK) Jesduvroq (daprodustat) to treat anaemia caused by chronic kidney disease (CKD) in adults who have been on dialysis for at least four months. It also achieved non-inferiority of MACE compared to ESA control, in the primary safety analysis of the ITT population.
The US Food and Drug Administration (FDA) has granted Emergency Use Authorizations (EUAs) for bivalent formulations of Moderna and Pfizer -BioNTech’s Covid-19 vaccines as boosters. The EUA amendments by the FDA are based on the totality of available data. In August, Moderna sought FDA EUA for the BA.4/BA.5 1 lineage.
The US Food and Drug Administration (FDA) has pushed quizartinib’s Prescription Drug User Fee Act (PDUFA) date in newly diagnosed FLT3-ITD positive acute myeloid leukemia (AML) for the review of updates of Risk Evaluation and Mitigation Strategies (REMS) included in the application. AML makes up 23.1% of total leukaemia cases globally.
The US Food and Drug Administration (FDA) has released a new draft guidance to further support the use of decentralised clinical trials (DCTs) for drugs, biologics and devices. This draft guidance builds on recommendations published by the agency in 2020.
a company committed to advancing surgical efficiency and safety, announced that they have received Safer Technologies Program (STeP) designation from the FDA for their surgical device StimSite. “Surgeons are constantly learning new techniques, either robotics or laparoscopic, and safety is always front and center.
On February 7, at a town hall organised to discuss clinical trial designs for gene therapies, FDA experts pushed pharma players to look for ways to establish clinical effectiveness despite the challenges in recruiting patients with rare diseases.
million in 2020, more than double the number in 2015, according to health data company IQVIA. The company decided there were too few reports of serious depression and suicidal behavior and not enough specifics about those cases to warrant more than “routine” monitoring of safety data. Annual U.S.
Adjuvanted vaccine Arexvy has been approved by the US Food and Drug Administration (FDA). Approval of GSK’s RSV vaccine The FDA granted approval of Arexvy to GlaxoSmithKline Biologicals. Now, the FDA has granted this milestone to GSK’s Arexvy. Could GSK offer the first RSV older adult vaccine?
In this article, we will explore the regulatory environment of pharmaceutical marketing and how companies are navigating it to balance patient safety and industry innovation. The United States Food and Drug Administration (FDA) is the primary regulatory agency that oversees the approval, promotion, and advertising of drugs.
It comes after Johnson & Johnson’s Janssen Biotech division signed up to access the bispecific antibody technology in 2020, without revealing any financial details. The biotech said earlier it planned to seek FDA approval to start clinical trials towards the end of 2022. 2 T cells, which have potent tumour-killing activity.
Sodium oxybate — which in the late 1980s was marketed to bodybuilders and then became known as GHB and criminally used as a date rape drug — has been sold under the brand name Xyrem after gaining FDA approval in 2002. In 2020, the FDA-approved indication was expanded to include those patients who suffer from cataplexy.
Adakveo (crizanlizumab) received conditional authorisation by the EC in October 2020. However, the STAND results did not suggest new safety concerns with crizanlizumab. Crizanlizumab is approved for use by the United States Food and Drug Administration (FDA).
Overall, the authors summarised that the enhanced sensitivity improves safety assessments for volatiles in medical devices. The paper discussed the detection of volatile extractables released during the clinical use of medical devices.
Treatment is then conducted as a clinical trial by the National Cancer Center Hospital to confirm its safety and efficacy. Under this system, patients can request the government for access to medical treatment using unapproved drugs not covered by insurance.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
Amgen has reported positive phase 3 results with its biosimilar version of AstraZeneca/Alexion’s blockbuster rare disease drug Soliris, setting up a regulatory filing with the FDA. The safety and immunogenicity profile of the biosimilar was also comparable to Alexion’s drug, said Amgen.
This is largely due to the FDA’s rigorous approach to the safety of microbiome therapeutics, which has manifested in clinical holds, resulting in delays that have dimmed the enthusiasm in the space in recent times. While the French MaaT Pharma has submitted further information to the FDA, its trial remains on hold. with placebo.
It refiled in the US, but got a complete response from the FDA in December with a request for more clinical information that has delayed the programme even further, and the CHMP’s decision will further undermine confidence in the drug’s prospects.
A previous October 2020 report concluded that longer-term safety and efficacy were needed to update health-benefit price benchmarks. The program was put on hold in December 2020 following a patient report, but this was lifted in April 2021. million, the think tank concluded in its updated evaluation. million ($1.8
An FDA advisory committee has recommended approval of Cidara Therapeutics’ once-weekly antifungal therapy rezafungin, setting it up to become the first new option for severe infections caused by Candida species in a decade. Melinta is privately-owned, after emerging from bankruptcy protection in 2020.
The company said it has started the processing for withdrawing the marketing authorisation for Blenrep (belantamab mafodotin) at the request of the FDA. “We will continue the DREAMM clinical trial programme and work with the US FDA on a path forward for this important treatment option for patients with multiple myeloma.”
4 The use of CPCA assessments allows for the assignment of scientifically justifiable higher acceptable intakes (AIs), without impacting patient safety. During August 2023 the FDA also updated its guidance for NDSRIs 7 and it was closely aligned (but not identical) to EMA’s evolving guidance. ng/day, compared to EMA which is 18 ng/day.
The US Food and Drug Administration (FDA) has accepted a priority review of the biologics license application (BLA) for Regeneron Pharmaceuticals ’ pozelimab to treat children and adults with ultra-rare CHAPLE disease. The post US FDA accepts Regeneron’s pozelimab BLA for priority review appeared first on Pharmaceutical Technology.
GSK, its consumer health spin-off Haleon and Sanofi could be on the hook for billions of dollars in potential damages over Zantac, a gastrointestinal drug pulled from the market in 2020 after it was linked to cases of cancer. Some voluntarily took their products off the market as soon as the NDMA finding was reported by the FDA.
In recent years, gene therapy has transitioned from a promising idea to a reality for patients, with many of the severe safety issues that emerged in early iterations of the technology being overcome. FDA backing. from 2021 to 2029.
That sets up a rolling Biologics License Application (BLA) with the FDA by the end of the year, setting Valneva on course to bring the first chikungunya vaccine to market anywhere in the world. Valneva estimates that the global market for vaccines against chikungunya could exceed $500 million annually by 2032.
2 Driven by the success of FMTs with clinical efficacy in several indications 3-6 and the first two FDA-approved donor-derived products on the market for recurrent Clostridioides difficile infections, 7,8 a new and promising trend is emerging that combines the ‘whole ecosystem’ of FMTs with rational consortia design. Microbiome.
Various uses soon became apparent including those for supply chain management, anti-counterfeiting, and to improve patient safety. The US Food and Drug Administration (FDA) has provided guidance for the use of RFID in the drug supply chain and to standardise the data format.
Additionally, when asked whether the FDA should consider a potential accelerated approval, panellists voted 15 to one against that regulatory pathway for the drug. Additionally, when asked whether the FDA should consider a potential accelerated approval, panellists voted 15 to one against that regulatory pathway for the drug.
Data announced at the European Lung Cancer Congress (ELCC) earlier this month revealed the long-term efficacy and safety of RYBREVANT ® (amivantamab) in patients with post-platinum EGFR Ex20ins-mutated advanced non-small cell lung cancer (NSCLC). 2020; 19(10): 2044–56. Personalized medicine at FDA. References 1. Garrido P et al.
The US Food and Drug Administration (FDA) subsequently developed a liquid chromatography – high resolution mass spectroscopy (LC-HRMS) method for the determination of NDMA in ranitidine. FDA assigned a provisional AI of 26.5 10 It is not clear whether FDA will adopt a similar t-AI. 2020 [cited 2023Jan]. 2020; 22(4): 89.
Less than a week before Ipsen’s palovarotene was due to go in front of an FDA advisory committee, the meeting has been called off following a request by the regulator for more data. The drug failed a futility test in a pivotal trial and was also placed under a partial clinical hold by the FDA while it probed the drug’s safety.
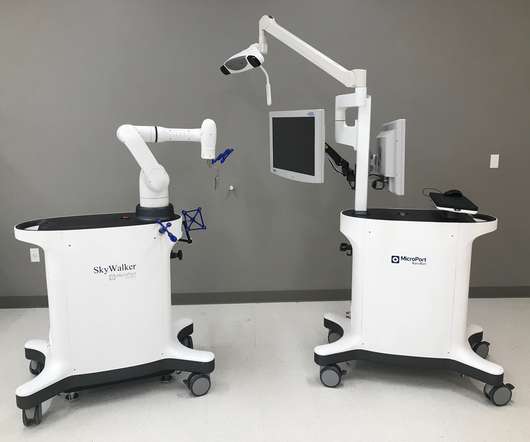
MicroPort Navibot has received 510(K) clearance from the Food and Drug Administration (FDA) in the United States for the SkyWalker System, the company’s first robot-assisted platform for orthopedic applications. The SkyWalker System has the technical advantages of precise operation and efficient coordination, while prioritizing safety.
This also occupies a large resource, given the US Food and Drug Administration (FDA) requirement for double plate checking using a second ‘independent’ person. The FDA is doing this through the publications of Guidance for Industry. This places a continued reliance upon plate reading and the limitations of human vision.
Miracor Medical SA (Miracor Medical) has announced the approval of an Investigational Device Exemption (IDE) from the FDA, enabling the company to initiate a pivotal study with its Pressure-controlled intermittent Coronary Sinus Occlusion (PiCSO) technology. About Miracor Medical. Press Release by: Miracor Medical. Are you hiring?
How feasible is it to establish a drug’s safety for use in a population that may be unwilling or unable to participate in clinical trials? Experts at a November 2021 Food and Drug Administration (FDA) meeting also wrestled with this theme of clinical trials and pregnancy. Medicine use during pregnancy: balancing risks.
To ensure the safety of medicines post-regulatory approval, a risk management plan (RMP) is established. As does the US Food and Drug Administration (FDA): REMS Assessment: Planning and Reporting Guidance for Industry Draft Guidance. What are risk minimisation methods? Before that she has been an independent scientific advisor.
If approved, gepotidacin (GSK2140944) could become the first drug in a new class of oral antibiotics for uncomplicated urinary tract infections (UTIs) in more than 20 years, according to the pharma group, which plans to submit the data to the FDA in the first half of next year.
2 The US Food and Drug Administration (FDA) has recently announced that it will control the quality of tobacco products, particularly e-cigarettes, more closely, to prevent avoidable contamination and help address “inconsistencies between product labelling and actual concentrations” in these products, potentially misleading customers.
We organize all of the trending information in your field so you don't have to. Join 8,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



































Let's personalize your content