This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The hurdles encountered in development and approval of a new drug can be overwhelming. 1 Consequently, the US Food and DrugAdministration (FDA) and other agencies are keen to see “accelerated development” programmes in areas where there is a significant unmet clinical need. billion, and rising. billion, and rising.
The US Food and DrugAdministration (FDA) has approved Rykindo ® (risperidone) for extended-release injectable suspension. The drug is indicated as a bi-weekly treatment for schizophrenia and as monotherapy or as adjunctive therapy to lithium or valproate for bipolar I disorder in adults.
Most psychedelic drugs are Schedule I controlled substances, which means that very strict legal and regulatory controls accompany their use” Some psychedelics originate in nature and have been used by Indigenous cultures for thousands of years; others are manipulated or manufactured. 1 Such restraints have stood solid for decades.
The ICH M10 guideline provides recommendations on the validation of bioanalytical assays for chemical and biological drugs and their metabolites in biological matrices. These concentration measurements are used as part of regulatory decisions regarding the safety and efficacy of medicinal products.
Rare disease drug development poses unique challenges that can be overcome by using real-world evidence (RWE). Another benefit is that fewer patients are required in clinical trials using HCs, thereby reducing time to trial completion and speeding drug approval. Patel et al.
Based on scientific knowledge and process understanding, manufacturers of drugs and drug substances have a broad awareness of potential impurities – including nitrosamine impurities – that could form during manufacturing or degradation. 1 Nitrosamines are not new or unknown impurities.
In this Q&A, Karen Pinachyan, Head of Medical Affairs Europe at CSL Behring summarises key considerations for gene therapy drug development and the ideal approach for alleviating economic strain when advancing these modern treatments. The US Food and DrugAdministration (FDA) requires a patient follow up period of at least 10-15 years.
percent of recalls recorded by the US Food and DrugAdministration (FDA) between 2012 and 2019. As a result of the rate of contamination events, simple and rapid detection of BCC in non-sterile pharmaceutical products is critical to ensure consumer safety. The research was published in Pathogens.
Of the 175 new drugs approved by the US Food and DrugAdministration (FDA) between 2016 and 2019, most were biologics. When the balloon reaches a sufficient pressure, this pushes the dissolvable needle into the intestinal wall, delivering the drug payload.”
The US Food and DrugAdministration (FDA) has accepted the biologics licence application (BLA) of Novartis subsidiary Sandoz for natalizumab, a proposed first-ever multiple sclerosis (MS) biosimilar to reference medicine Tysabri. Natalizumab is developed by Sandoz’s collaboration partner Polpharma Biologics.
This recent decision by the EC follows approval of Tyruko ® (natalizumab) by the US Food and DrugAdministration (FDA) last month for the same indication. Sandoz entered into a global commercialization agreement for biosimilar natalizumab with Polpharma Biologics in 2019.
In June last year, Eli Lilly submitted a new drug application to the National Medical Products Administration (NMPA) of China for Galcanezumab to prevent episodic migraine in adult patients. In September 2018, the US Food and DrugAdministration (FDA) approved Galcanezumab as the preventive treatment of migraine in adults.
Notably, Lupkynis is the first oral therapy approved by the US Food and DrugAdministration (FDA) and EC for the treatment of active LN. Key opinion leaders (KOLs) interviewed by GlobalData believe this may have significant impact on patients as the less-invasive route of administration will be preferred by many.
IL-2 therapy is associated with safety concerns, namely the adverse event capillary leak syndrome, which often leads to hospitalisations and thus limits its clinical use. Protein engineering comes with several challenges, including maintaining stability and immunogenicity of the novel protein and creating an inherently active drug.
Drug developers often face a Catch-22 regarding clinical trials and pregnancy. How feasible is it to establish a drug’ssafety for use in a population that may be unwilling or unable to participate in clinical trials? Panelists were asked to discuss the potential use of the drug during pregnancy.
The validation of computerized systems should be focused on ‘intended use’ and scaled commensurate with the risks to patient safety, product quality and data integrity” The focus until now has too often been on compliance and not on quality. Takeaways from ISPE’s GAMP 5 Second Edition update.
In 2019, Daiichi Sankyo entered a global development and commercialisation agreement with AstraZeneca for Daiichi Sankyo’s lead antibody-drug conjugate (ADC), Enhertu (trastuzumab deruxtecan), in a deal worth $6.9bn. overall response rate (ORR), with a median duration of response (DOR) of 8.7
Food and DrugAdministration (FDA) has approved MRI conditional labeling for the Evoke ® System, the first and only precision, dose-control spinal cord stimulation (SCS) therapy powered by SmartLoop technology. Specific scan conditions and safety information are provided in the Evoke ® SCS System MRI Guidelines manual.
The US Food and DrugAdministration (FDA) subsequently developed a liquid chromatography – high resolution mass spectroscopy (LC-HRMS) method for the determination of NDMA in ranitidine. 1,2 The second occurrence of NDMA arising from degradation was in metformin drug product.
Recent policy and regulatory moves have begun to sketch out a framework for rare disease drug approvals in the country, but obstacles to approvals and patient access remain. The list, designed to serve as a reference point for prioritized drug review and evaluation policies, included 121 rare diseases. [8]. 10] [11] [12].
However, numbers are still much higher than 2019, indicating the use of telemedicine is set to remain post-Covid. Lower DCT usage for Phase I studies may be accounted for due to the heavier focus on safety and efficacy, and increased monitoring of trial participants in-person.
The US Food and DrugAdministration (FDA) has pushed quizartinib’s Prescription Drug User Fee Act (PDUFA) date in newly diagnosed FLT3-ITD positive acute myeloid leukemia (AML) for the review of updates of Risk Evaluation and Mitigation Strategies (REMS) included in the application. Quizartinib had a 11.3%
AstraZeneca and Daiichi Sankyo ’s Enhertu (trastuzumab deruxtecan) has received expanded approval from the US Food and DrugAdministration (FDA) to treat adults with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer. Enhertu-treated subjects had a median overall survival (OS) of 23.4
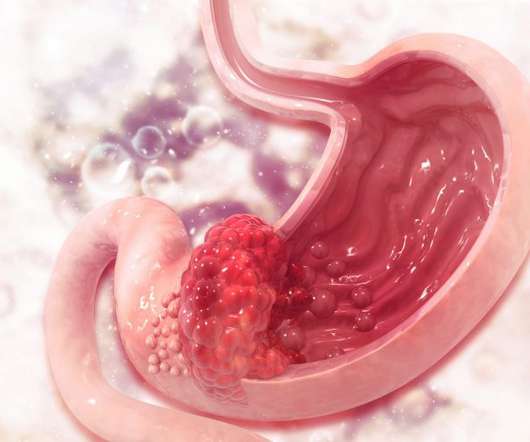
Several lifestyle factors have been shown to increase the risk of developing stomach cancer, including alcohol consumption, smoking and consuming foods preserved by salts. There is an urgent need for more effective chemotherapy treatments with less severe toxicity and side effects. New hope for stomach cancer patients.
FAKE MEDICINES are not a new problem, and pose a tremendous threat to patient safety and public health in society regardless of a country’s economic status. The prevalence of fake drugs is a continually growing problem worldwide. Fake drugs kill more than 250,000 children a year, doctors warn. 2019; Available from: [link].
The following year, the US Congress passed the 1938 Federal Food, Drug and Cosmetic (FD&C) Act, which mandated safety assessments prior to the release of any new drug. Elixirs, diluents, and the passage of the 1938 Federal Food, Drug and Cosmetic Act. THE TRUTH PILL: The Myth of Drug Regulation in India.
1 Some five years after the initial NDMA (N-nitrosodimethylamine) contamination issue 2 initially affecting valsartan drug substance, then other active pharmaceutical ingredients (APIs), eg, sartans, ranitidine, metformin, etc; the toxic short alkyl chain N-nitrosamine issue appears, if not resolved, then well on the way to resolution.
The US Food and DrugAdministration (FDA) granted Eli Lilly’s Lartruvo (olaratumab) accelerated approval in 2016 for soft tissue sarcoma (STS). The US Food and DrugAdministration (FDA) granted Eli Lilly’s Lartruvo (olaratumab) accelerated approval in 2016 for soft tissue sarcoma (STS).
The success of mRNA vaccines against SARS-CoV-2 has quickly catapulted mRNA therapeutics as a disruptive, expanding drug category” The term ‘mRNA’ has become commonplace globally. The success of mRNA vaccines against SARS-CoV-2 has quickly catapulted mRNA therapeutics as a disruptive, expanding drug category.
Limitations of monoclonal antibody therapies Regulatory approvals from the US Food and DrugAdministration (FDA) for aducanumab and lecanemab – and likely very soon for donanemab also – opened a route for different therapeutic modalities and other relevant disease targets, such as tau.
The US Food and DrugAdministration (FDA) has sent a warning letter to KVK-Techs drug manufacturing facility following an inspection in April 2019. Although the particles were filtered out, the company failed to thoroughly examine their origin, nature, and potential impact on drug quality.
Opdivo is a programmed cell death receptor-1 (PD-1) inhibitor approved by the US Food and DrugAdministration (FDA) for the treatment of several cancers. months, following an ESMO 2019 presentation on data from the same study. This update involved a longer follow-up period of 30.4
One element that project managers often forget or underestimate is the environmental, health and safety (EHS) tasks” Operating PAT from hardware to software, in a manufacturing suite, would not be successful without training and competency development to track training status in a learning management system for individuals.
We organize all of the trending information in your field so you don't have to. Join 8,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






























Let's personalize your content